DIAGNOSTIC MEDICAL MICROBIOLOGY
PRINCIPLES OF DIAGNOSTIC MEDICAL MICROBIOLOGY CONTD...
IDENTIFYING BACTERIA USING 16S RRNA
The 16S rRNA of each species of bacteria has stable (conserved) portions of the sequence. Many copies are present in each organism. Labeled probes specific for the 16S rRNA of a species are added, and the amount of label on the double-stranded hybrid is measured. This technique is widely used for the rapid identification of many organisms. Examples include the most common and important Mycobacterium species, C immitis, Histoplasma capsulatum, and others. Portions of the 16S rRNA are conserved across many species of microorganisms. Amplifying the 16S rRNA using primers to these conserved regions allows isolation and sequencing of the variable regions of the molecules. These variable sequences are genus- or species-specific markers that allow identification of microorganisms. Pathogens that are difficult or impossible to culture in the laboratory have been identified using this technique.One example is Tropheryma whipplei, the cause of Whipple disease. Molecular diagnostic assays that use amplification of nucleic acid have become widely used and are evolving rapidly . These amplification systems fall into several basic categories as outlined below.
TARGET AMPLIFICATION SYSTEMS
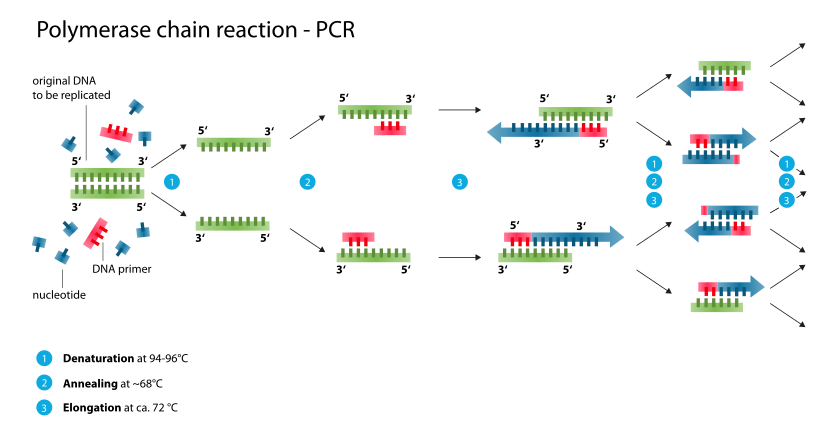
In these assays, the target DNA or RNA is amplified many times. The polymerase chain reaction (PCR) is
used to amplify extremely small amounts of specific DNA present in a clinical specimen, making it possible to detect what were initially minute amounts of the DNA. PCR uses a thermostable DNA polymerase to produce a twofold amplification of target DNA with each temperature cycle. Conventional PCR utilizes three sequential reactions—denaturation, annealing, and primer extension—as follows. The DNA extracted from the clinical specimen along with sequence-specific oligonucleotide primers, nucleotides, thermostable DNA polymerase,and buffer are heated to 90–95°C to denature (separate) the two strands of the target DNA. The temperature in the reaction is lowered, usually to 45–60°C depending upon the primers, to allow annealing of the primers to the target DNA. Each primer is then extended by the thermostable DNA polymerase by adding nucleotides complementary to the target DNA yielding the twofold amplification. The cycle is then repeated 30–40 times to yield amplification of the target DNA segment by as much as 10^5 - 10^6 fold. The amplified segment often can be seen in an electrophoretic gel or detected by Southern blot analysis using labeled DNA probes specific for the segment or by a variety of proprietary commercial techniques. PCR can also be performed on RNA targets, which is called reverse transcriptase PCR. The enzyme reverse transcriptase is used to transcribe the RNA into complementary DNA for amplification. PCR assays are available commercially for identification of a broad range of bacterial and viral pathogens such as Chlamydia trachomatis, N gonorrhoeae, M tuberculosis, cytomegalovirus, enteroviruses, and many others.An assay is available for HIV -1 viral load testing also. There are many other "in-house" PCRs that have been developed by individual laboratories to diagnose infections. Such assays are the tests of choice to diagnose many infections—especially when traditional culture and antigen detection techniques do not work well. Examples include testing of cerebrospinal fluid for herpes simplex virus to diagnose herpes encephalitis and testing of nasopharyngeal wash fluid to diagnose B pertussis infection (whooping cough). A major consideration for laboratories that perform PCR assays is to prevent contamination of reagents or specimens with target DNA from the environment, which can obscure the distinction between truly positive results and falsely positive ones because of the contamination.
PROBE AMPLIFICATION SYSTEMS
The ligase chain reaction (LCR) is an amplification system different from PCR. LCR uses thermostable DNA polymerase and thermostable DNA ligase. LCR uses four oligonucleotide probes of 20–24 bases each. Each pair of oligonucleotides is designed to bind to the denatured target DNA only a few bases apart. The oligonucleotides are mixed with extracted target DNA from the specimen and other reagents and then heated to denature the target DNA. The reaction is then cooled to allow binding of the oligonucleotide probes to the target DNA. The short gap between the two probes is filled in by the DNA polymerase and linked by the DNA ligase, yielding double-stranded DNA molecules 40–50 bp in length. The cycle is repeated 30–40 times, yielding a large number of DNA molecules. This commercially available system includes automated detection of the amplified DNA. It can be used to detect C trachomatis and N gonorrhoeae. It is available only outside of the United States.
SIGNAL AMPLIFICATION TECHNIQUES
These assays strengthen the signal by amplifying the label (eg, fluorochromes, enzymes) that is attached to the target nucleic acid. The branched DNA (bDNA) system has a series of primary probes and a branched
secondary probe labeled with enzyme. Multiple oligonucleotide probes specific for the target RNA (or DNA) are fixed to a solid surface such as a microdilution tray . These are the capture probes. The prepared specimen is added, and the RNA molecules are attached to the capture probes on the microdilution tray . Additional target probes bind to the target but not to the tray . The enzyme-labeled bDNA amplifier probes are added and attach to the target probes. A chemiluminescent substrate is added, and light emitted is measured to quantitate the amount of target RNA present. Examples of the use of this type of assay include the quantitative measurement of HIV -1, hepatitis C virus, and hepatitis B virus.
AMPLIFICATION METHODS: NON–PCR-BASED
The transcription-mediated amplification (TMA) and the nucleic acid sequence-based amplification
(NASBA) systems amplify large quantities of RNA in isothermal assays that coordinately use the enzymes
reverse transcriptase, RNase H, and RNA polymerase. An oligonucleotide primer containing the RNA
polymerase promoter is allowed to bind to the RNA target. The reverse transcriptase makes a single-stranded cDNA copy of the RNA. The RNase H destroys the RNA of the RNA -cDNA hybrid, and a second primer anneals to the segment of cDNA. The DNA -dependent DNA polymerase activity of reverse transcriptase extends the DNA from the second primer , producing a double-stranded DNA copy , with intact RNA polymerase. The RNA polymerase then produces many copies of the single-stranded RNA. Detection of C trachomatis, N gonorrhoeae,and M tuberculosis and quantitation of HIV -1 load are examples of the use of these types of assays. Strand displacement assays (SDA) are isothermal amplification assays that employ use of restrictive endonuclease and DNA polymerase.
REAL-TIME PCR
Technologic advances, which have lead to "real-time amplification," have streamlined nucleic acid amplification platforms,improved the sensitivity of amplification tests, and have drastically reduced the potential for contamination. Real-time instruments have replaced the solid block used in conventional thermocyclers with fans that allow more rapid PCR cycling. Dramatic improvements in the chemistry of nucleic acid amplification reactions have resulted in homogeneous reaction mixtures in which fluorogenic compounds are present in the same reaction tube in which the amplification occurs. A variety of fluorogenic molecules are used. These include nonspecific dyes such as SYBR green, which binds to the minor groove of double-stranded DNA, and amplicon specific detection methods using fluorescently labeled oligonucleotide probes, which fall into three categories:
TaqMan or hydrolysis probes; fluorescence energy transfer (FRET) probes; and molecular beacons. All of the methods allow for measurement of fluorescence with each amplification cycle, that is, "real-time" assessment of results. Since the reaction tube does not need to be opened to analyze the PCR products on a gel, there is much less risk of amplicon carry-over to the next reaction.
THE IMPORTANCE OF NORMAL BACTERIAL & FUNGAL FLORA
Organisms such as M tuberculosis, Salmonella typhi, and Brucella species are considered pathogens whenever they are found in patients. However , many infections are caused by organisms that are permanent or transient members of the normal flora. For example, Escherichia coli is part of the normal gastrointestinal flora and is also the most common cause of urinary tract infections. Similarly , the vast majority of mixed bacterial infections with anaerobes are caused by organisms that are members of the normal flora. The relative numbers of specific organisms found in a culture are important when members of the normal flora are the cause of infection.When numerous gram-negative rods of species such as Klebsiella pneumoniae are found mixed with a few normal nasopharyngeal bacteria in a sputum culture, the gram-negative rods are strongly suspect as the cause of pneumonia because large numbers of gram-negative rods are not normally found in sputum or in the nasopharyngeal flora; the organisms should be identified and reported. In contrast,abdominal abscesses commonly contain a normal distribution of aerobic, facultatively anaerobic, and obligately anaerobic organisms representative of the gastrointestinal flora. In such cases, identification of all species present is not warranted; instead, it is appropriate to report "normal gastrointestinal flora." Yeasts in small numbers are commonly part of the normal microbial flora. However , other fungi are not normally present and therefore should be identified and reported. Viruses usually are not part of the normal flora as detected in diagnostic microbiology laboratories. However , some latent viruses, eg, herpes simplex, or live vaccine viruses such as poliovirus occasionally appear in cultures for viruses. In some parts of the world,stool specimens commonly yield evidence of parasitic infection. In such cases, it is the relative number of parasites correlated with the clinical presentation that is important.
CITED BY ANIL BHUJEL
BSC MICROBIOLOGY, TU.
MICROBIOLOGY STUDENT AT PBPC, NAYABAZZAR-9, POKHARA.
SOME SUGGESTED REFERENCES:
www.ncbi.nlm.nih.gov › ... › J Clin Microbiol › v.45(9); Sep 2007
en.wikipedia.org/wiki/16S_ribosomal_RNA
biotech.bio5.org/sites/default/files/pdf/16s%20rDNA%20SeqAnal.pdf
link.springer.com/chapter/10.1007%2F0-387-32892-0_20
amrita.vlab.co.in/?sub=3&brch=76&sim=1421&cnt=1
www.austincc.edu/.../mdfund_unit9Chapter7NucleicAcidAmplificationT...
www.genengnews.com/.../targeted-amplification-system.../81245623/?...
www.ncbi.nlm.nih.gov/pubmed/1370001
https://www.inkling.com/read/.../signal-amplification-methods
en.wikipedia.org/wiki/Polymerase_chain_reaction.
books.google.com.np/books?isbn=1402062419
www.mdpi.com/2079-6374/3/1/18/pdf
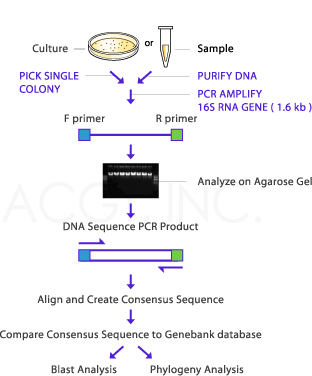



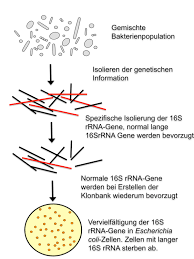




IDENTIFYING BACTERIA USING 16S RRNA
The 16S rRNA of each species of bacteria has stable (conserved) portions of the sequence. Many copies are present in each organism. Labeled probes specific for the 16S rRNA of a species are added, and the amount of label on the double-stranded hybrid is measured. This technique is widely used for the rapid identification of many organisms. Examples include the most common and important Mycobacterium species, C immitis, Histoplasma capsulatum, and others. Portions of the 16S rRNA are conserved across many species of microorganisms. Amplifying the 16S rRNA using primers to these conserved regions allows isolation and sequencing of the variable regions of the molecules. These variable sequences are genus- or species-specific markers that allow identification of microorganisms. Pathogens that are difficult or impossible to culture in the laboratory have been identified using this technique.One example is Tropheryma whipplei, the cause of Whipple disease. Molecular diagnostic assays that use amplification of nucleic acid have become widely used and are evolving rapidly . These amplification systems fall into several basic categories as outlined below.
TARGET AMPLIFICATION SYSTEMS
In these assays, the target DNA or RNA is amplified many times. The polymerase chain reaction (PCR) is
used to amplify extremely small amounts of specific DNA present in a clinical specimen, making it possible to detect what were initially minute amounts of the DNA. PCR uses a thermostable DNA polymerase to produce a twofold amplification of target DNA with each temperature cycle. Conventional PCR utilizes three sequential reactions—denaturation, annealing, and primer extension—as follows. The DNA extracted from the clinical specimen along with sequence-specific oligonucleotide primers, nucleotides, thermostable DNA polymerase,and buffer are heated to 90–95°C to denature (separate) the two strands of the target DNA. The temperature in the reaction is lowered, usually to 45–60°C depending upon the primers, to allow annealing of the primers to the target DNA. Each primer is then extended by the thermostable DNA polymerase by adding nucleotides complementary to the target DNA yielding the twofold amplification. The cycle is then repeated 30–40 times to yield amplification of the target DNA segment by as much as 10^5 - 10^6 fold. The amplified segment often can be seen in an electrophoretic gel or detected by Southern blot analysis using labeled DNA probes specific for the segment or by a variety of proprietary commercial techniques. PCR can also be performed on RNA targets, which is called reverse transcriptase PCR. The enzyme reverse transcriptase is used to transcribe the RNA into complementary DNA for amplification. PCR assays are available commercially for identification of a broad range of bacterial and viral pathogens such as Chlamydia trachomatis, N gonorrhoeae, M tuberculosis, cytomegalovirus, enteroviruses, and many others.An assay is available for HIV -1 viral load testing also. There are many other "in-house" PCRs that have been developed by individual laboratories to diagnose infections. Such assays are the tests of choice to diagnose many infections—especially when traditional culture and antigen detection techniques do not work well. Examples include testing of cerebrospinal fluid for herpes simplex virus to diagnose herpes encephalitis and testing of nasopharyngeal wash fluid to diagnose B pertussis infection (whooping cough). A major consideration for laboratories that perform PCR assays is to prevent contamination of reagents or specimens with target DNA from the environment, which can obscure the distinction between truly positive results and falsely positive ones because of the contamination.
PROBE AMPLIFICATION SYSTEMS
The ligase chain reaction (LCR) is an amplification system different from PCR. LCR uses thermostable DNA polymerase and thermostable DNA ligase. LCR uses four oligonucleotide probes of 20–24 bases each. Each pair of oligonucleotides is designed to bind to the denatured target DNA only a few bases apart. The oligonucleotides are mixed with extracted target DNA from the specimen and other reagents and then heated to denature the target DNA. The reaction is then cooled to allow binding of the oligonucleotide probes to the target DNA. The short gap between the two probes is filled in by the DNA polymerase and linked by the DNA ligase, yielding double-stranded DNA molecules 40–50 bp in length. The cycle is repeated 30–40 times, yielding a large number of DNA molecules. This commercially available system includes automated detection of the amplified DNA. It can be used to detect C trachomatis and N gonorrhoeae. It is available only outside of the United States.
SIGNAL AMPLIFICATION TECHNIQUES
These assays strengthen the signal by amplifying the label (eg, fluorochromes, enzymes) that is attached to the target nucleic acid. The branched DNA (bDNA) system has a series of primary probes and a branched
secondary probe labeled with enzyme. Multiple oligonucleotide probes specific for the target RNA (or DNA) are fixed to a solid surface such as a microdilution tray . These are the capture probes. The prepared specimen is added, and the RNA molecules are attached to the capture probes on the microdilution tray . Additional target probes bind to the target but not to the tray . The enzyme-labeled bDNA amplifier probes are added and attach to the target probes. A chemiluminescent substrate is added, and light emitted is measured to quantitate the amount of target RNA present. Examples of the use of this type of assay include the quantitative measurement of HIV -1, hepatitis C virus, and hepatitis B virus.
AMPLIFICATION METHODS: NON–PCR-BASED
The transcription-mediated amplification (TMA) and the nucleic acid sequence-based amplification
(NASBA) systems amplify large quantities of RNA in isothermal assays that coordinately use the enzymes
reverse transcriptase, RNase H, and RNA polymerase. An oligonucleotide primer containing the RNA
polymerase promoter is allowed to bind to the RNA target. The reverse transcriptase makes a single-stranded cDNA copy of the RNA. The RNase H destroys the RNA of the RNA -cDNA hybrid, and a second primer anneals to the segment of cDNA. The DNA -dependent DNA polymerase activity of reverse transcriptase extends the DNA from the second primer , producing a double-stranded DNA copy , with intact RNA polymerase. The RNA polymerase then produces many copies of the single-stranded RNA. Detection of C trachomatis, N gonorrhoeae,and M tuberculosis and quantitation of HIV -1 load are examples of the use of these types of assays. Strand displacement assays (SDA) are isothermal amplification assays that employ use of restrictive endonuclease and DNA polymerase.
REAL-TIME PCR
Technologic advances, which have lead to "real-time amplification," have streamlined nucleic acid amplification platforms,improved the sensitivity of amplification tests, and have drastically reduced the potential for contamination. Real-time instruments have replaced the solid block used in conventional thermocyclers with fans that allow more rapid PCR cycling. Dramatic improvements in the chemistry of nucleic acid amplification reactions have resulted in homogeneous reaction mixtures in which fluorogenic compounds are present in the same reaction tube in which the amplification occurs. A variety of fluorogenic molecules are used. These include nonspecific dyes such as SYBR green, which binds to the minor groove of double-stranded DNA, and amplicon specific detection methods using fluorescently labeled oligonucleotide probes, which fall into three categories:
TaqMan or hydrolysis probes; fluorescence energy transfer (FRET) probes; and molecular beacons. All of the methods allow for measurement of fluorescence with each amplification cycle, that is, "real-time" assessment of results. Since the reaction tube does not need to be opened to analyze the PCR products on a gel, there is much less risk of amplicon carry-over to the next reaction.
THE IMPORTANCE OF NORMAL BACTERIAL & FUNGAL FLORA
Organisms such as M tuberculosis, Salmonella typhi, and Brucella species are considered pathogens whenever they are found in patients. However , many infections are caused by organisms that are permanent or transient members of the normal flora. For example, Escherichia coli is part of the normal gastrointestinal flora and is also the most common cause of urinary tract infections. Similarly , the vast majority of mixed bacterial infections with anaerobes are caused by organisms that are members of the normal flora. The relative numbers of specific organisms found in a culture are important when members of the normal flora are the cause of infection.When numerous gram-negative rods of species such as Klebsiella pneumoniae are found mixed with a few normal nasopharyngeal bacteria in a sputum culture, the gram-negative rods are strongly suspect as the cause of pneumonia because large numbers of gram-negative rods are not normally found in sputum or in the nasopharyngeal flora; the organisms should be identified and reported. In contrast,abdominal abscesses commonly contain a normal distribution of aerobic, facultatively anaerobic, and obligately anaerobic organisms representative of the gastrointestinal flora. In such cases, identification of all species present is not warranted; instead, it is appropriate to report "normal gastrointestinal flora." Yeasts in small numbers are commonly part of the normal microbial flora. However , other fungi are not normally present and therefore should be identified and reported. Viruses usually are not part of the normal flora as detected in diagnostic microbiology laboratories. However , some latent viruses, eg, herpes simplex, or live vaccine viruses such as poliovirus occasionally appear in cultures for viruses. In some parts of the world,stool specimens commonly yield evidence of parasitic infection. In such cases, it is the relative number of parasites correlated with the clinical presentation that is important.
CITED BY ANIL BHUJEL
BSC MICROBIOLOGY, TU.
MICROBIOLOGY STUDENT AT PBPC, NAYABAZZAR-9, POKHARA.
SOME SUGGESTED REFERENCES:
www.ncbi.nlm.nih.gov › ... › J Clin Microbiol › v.45(9); Sep 2007
en.wikipedia.org/wiki/16S_ribosomal_RNA
biotech.bio5.org/sites/default/files/pdf/16s%20rDNA%20SeqAnal.pdf
link.springer.com/chapter/10.1007%2F0-387-32892-0_20
amrita.vlab.co.in/?sub=3&brch=76&sim=1421&cnt=1
www.austincc.edu/.../mdfund_unit9Chapter7NucleicAcidAmplificationT...
www.genengnews.com/.../targeted-amplification-system.../81245623/?...
www.ncbi.nlm.nih.gov/pubmed/1370001
https://www.inkling.com/read/.../signal-amplification-methods
en.wikipedia.org/wiki/Polymerase_chain_reaction.
books.google.com.np/books?isbn=1402062419
www.mdpi.com/2079-6374/3/1/18/pdf


Comments