ASPECTS OF MOLECULAR BIOLOGY & BIOINFORMATICS RELEVANCE IN INDUSTRIAL MICROBIOLOGY & BIOTECHNOLOGY CONTD..
METAGENOMICS:
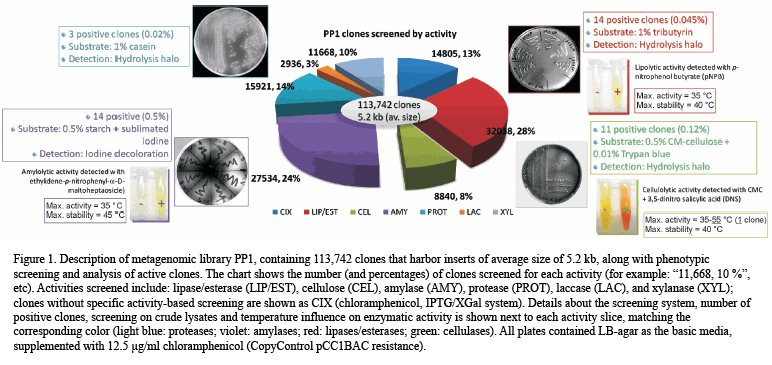
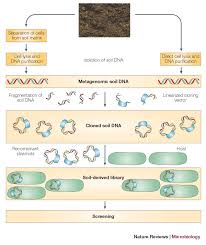
Metagenomics is the genomic analysis of the collective genome of an assemblage of
organisms or ‘metagenome.’Metagenomics describes the functional and sequence-based
analysis of the collective microbial genomes contained in an environmental sample.
Other terms have been used to describe the same method, including
environmental DNA libraries, zoolibraries, soil DNA libraries, eDNA libraries,
recombinant environmental libraries, whole genome treasures, community genome,
whole genome shotgun sequencing. The definition applied here excludes studies that
use PCR to amplify gene cassettes or random PCR primers to access genes of interest since these methods do not provide genomic information beyond the genes that are amplified.
Uncultured microorganisms comprise the majority of the planet earth’s biological
diversity. In many environments, as many as 99% of the microorganisms cannot be
cultured by standard techniques, and the uncultured fraction includes diverse
organisms that are only distantly related to the cultured ones. Therefore, culture independent methods are essential to understand the genetic diversity, population
structure, and ecological roles of the majority of microorganisms in a given
environmental situation. Metagenomics, or the culture-independent genomic analysis of
an assemblage of microorganisms, has potential to answer fundamental questions in
microbial ecology. It can also be applied to determining organisms which may be
important in a new industrial process still under study. Several markers have been used
in metagenomics, including 16S mRNA, and the genes encoding DNA polymerases,
because these are highly conserved (i.e., because they remain relatively unchanged in
many groups). The marker most commonly used however is the sequence of 16S mRNA.
Its potential application in biotechnology and industrial microbiology is that it can
facilitate the identification of uncultured organisms whose role in a multi-organism
environment such as sewage or the degradation of a recalcitrant chemical soil may be
hampered because of the inability to culture the organism. Indeed a method has been
patented for isolating organisms of pharmaceutical importance from uncultured
organisms in the environment.
 NATURE OF BIOINFORMATICS:
NATURE OF BIOINFORMATICS:
Bioinformatics is a new and evolving science and may be defined as the use of computers
to store, compare, retrieve, analyze, predict, or simulate the composition or the structure
of the genetic macromolecules, DNA and RNA and their major product, proteins.
Important research efforts in bioinformatics include sequence alignment, gene finding,
genome assembly, protein structure alignment, protein structure prediction, prediction of
gene expression and protein-protein interactions, and the modeling of evolution.
Bioinformatics uses mathematical tools to extract useful information from a variety of
data produced by high-throughput biological techniques. Examples of successful
extraction of orderly information from a ‘forest’of seemingly chaotic information include
the assembly of high-quality DNA sequences from fragmentary ‘shotgun’DNA
sequencing, and the prediction of gene regulation with data from mRNA microarrays or
mass spectrometry.
The increased role in recent times of bioinformatics in biotechnology is due to a vast
increase in computation speed and memory storage capability, making it possible to
undertake problems unthinkable without the aid of computers. Such problems include
large-scale sequencing of genomes and management of large integrated databases over
the Internet. This improved computational capability integrated with large-scale
miniaturization of biochemical techniques such as PCR, BAC, gel electrophoresis, and
microarray chips has delivered enormous amount of genomic and proteomic data to the
researchers. The result is an explosion of data on the genome and proteome analysis
leading to many new discoveries and tools that are not possible in wet-laboratory
experiments. Thus, hundreds of microbial genomes and many eukaryotic genomes
including a cleaner draft of human genome have been sequenced raising the expectation
of better control of microorganisms. Bioinformatics has been used in the following four
areas:
a. genomics –sequencing and comparative study of genomes to identify gene and
genome functionality;
b. proteomics –identification and characterization of protein related properties and
reconstruction of metabolic and regulatory pathways;
c. cell visualization and simulation to study and model cell behavior; and
d. application to the development of drugs and anti-microbial agents.
The potential gains especially following from sequencing of the human genome and
many microorganisms are greater understanding of the genetics of microorganisms and
their subsequent improved control leading to better diagnosis of the diseases through the
use of protein biomarkers, protection against diseases using cost effective vaccines and rational drug design, and improvement in agricultural quality and quantity.
Some Contributions of Bioinformatics to Biotechnology:
Some contributions made by bioinformatics to biotechnology include automatic genome
sequencing, automatic identification of genes, identification of gene function, predicting
the 3D structure modeling and pair-wise comparison of genomes.
i. Automatic genome sequencing
The major contribution of the bioinformatics in genome sequencing has been in the: (i)
development of automated sequencing techniques that integrate the PCR or BAC based
amplification, 2D gel electrophoresis and automated reading of nucleotides, (ii) joining
the sequences of smaller fragments (contigs) together to form a complete genome
sequence, and (iii) the prediction of promoters and protein coding regions of the genome.
PCR (Polymerase Chain Reaction) or BAC (Bacterial Artificial Chromosome)-based
amplification techniques derive limited size fragments of a genome. The available
fragment sequences suffer from nucleotide reading errors, repeats –very small and very
similar fragments that fit in two or more parts of a genome, and chimera –two different
parts of the genome or artifacts caused by contamination that join end-to-end giving a
artifactual fragment. Generating multiple copies of the fragments, aligning the fragments,
and using the majority voting at the same nucleotide positions solve the nucleotide
reading error problem. Multiple experimental copies are needed to establish repeats and
chimeras. Chimeras and repeats are removed before the final assembly of the genome fragments. Using mathematical models, the fragments are joined. To join contigs, the
fragments with larger nucleotide sequence overlap are joined first.
ii. Automated Identification of Genes
After the contigs are joined, the next issue is to identify the protein coding regions or ORFs
(open reading frames) in the genomes. The identification of ORFs is based on the
principles described earlier. The two programs which are used are GLIMMER and
GenBank.
iii. Identifying gene function: searching and alignment
After identifying the ORFs, the next step is to annotate the genes with proper structure
and function. The function of the gene has been identified using popular sequence search
and pair-wise gene alignment techniques. The four most popular algorithms used for
functional annotation of the genes are BLAST, BLOSUM, ClustalX, and SMART
iv. Three-dimensional (3D) structure modeling
A protein may exist under one or more conformational states depending upon its
interaction with other proteins. Under a stable conformational state certain regions of the
protein are exposed for protein-protein or protein-DNA interactions. Since the function is
also dependent upon exposed active sites, protein function can be predicted by matching
the 3D structure of an unknown protein with the 3D structure of a known protein. With bioinformatics it is possible to predict the possible conformations of the protein coded for
by a gene and therefore the function of the protein.
v. Pair-wise genome comparison
After the identification of gene-functions, a natural step is to perform pair-wise genome
comparisons. Pair-wise genome comparison of a genome against itself provides the
details of paralogous genes –duplicated genes that have similar sequence with some
variation in function. Pairwise genome comparisons of a genome against other genomes
have been used to identify a wealth of information such as orthologous genes –
functionally equivalent genes diverged in two genomes due to speciation, different types
of gene-groups –adjacent genes that are constrained to occur in close proximity due to
their involvement in some common higher level function, lateral gene-transfer –gene
transfer from a microorganism that is evolutionary distant, gene-fusion/gene-fission,
gene-group duplication, gene-duplication, and difference analysis to identify genes
specific to a group of genomes such as pathogens, and conserved genes.
In conclusion, despite the recent emergence of bioinformatics it is already making big
impacts on biotechnology. Except for the availability of bioinformatics techniques, the
vast amount of data generated by genome sequencing projects would be unmanageable
and would not be interpreted due to the lack of expert manpower and due to the
prohibitive cost of sustaining such an effort. In the last decade bioinformatics has silently
filled in the role of cost effective data analysis. This has quickened the pace of discoveries,
the drug and vaccine design, and the design of anti-microbial agents. The major impact of
bioinformatics in microbiology and biotechnology has been in automating microbial
genome sequencing, the development of integrated databases over the Internet, and
analysis of genomes to understand gene and genome function. Programs exist for
comparing gene-pair alignments, which become the first steps to derive the gene-function
and the functionality of genomes. Using bioinformatics techniques it is now possible to
compare genomes so as to (i) identify conserved function within a genome family; (ii)
identify specific genes in a group of genomes; and (iii) model 3D structures of proteins
and docking of biochemical compounds and receptors. These have direct impact in the
development of antimicrobial agents, vaccines, and rational drug design.
SUGGESTED READINGS & REFERENCES:
1)Madigan, M., Martinko, J.M. 2006. Brock Biology of Microorganisms 11th ed. Pearson Prentice Hall,
Upper Saddle River, USA.
2) Dorrell, N., Champion, O.L., Wren, B.W. 2002. Application of DNA Microarray for Comparative
and Evolutionary Genomics In: Methods in Microbiology. Vol 33, Academic Press
Amsterdam; the Netherlands pp. 83–99.
3) Riesenfeld, C.S., Schloss, P.D., Handelsman, 2004. Metagenomics: Genomic Analysis of Microbial
Communities. Annual Review of Genetics 38, 525-52.
CITED BY KAMAL SINGH KHADKA
Some Useful Urls:
en.wikipedia.org/wiki/Metagenomics
www.nature.com/nrmicro/focus/metagenomics/
www.cbcb.umd.edu/research/metagenomics.shtml
www.nature.com/ncomms/archive/subject/npg_subject_48/index.html
www.nature.com › Journal home › Archive › Technologies
www.nature.com/news/archive/keyword/bioinformatics.html
www.ebi.ac.uk/luscombe/docs/imia_review.pdf
Metagenomics is the genomic analysis of the collective genome of an assemblage of
organisms or ‘metagenome.’Metagenomics describes the functional and sequence-based
analysis of the collective microbial genomes contained in an environmental sample.
Other terms have been used to describe the same method, including
environmental DNA libraries, zoolibraries, soil DNA libraries, eDNA libraries,
recombinant environmental libraries, whole genome treasures, community genome,
whole genome shotgun sequencing. The definition applied here excludes studies that
use PCR to amplify gene cassettes or random PCR primers to access genes of interest since these methods do not provide genomic information beyond the genes that are amplified.
Many environments have been focus of metagenomics, including the soil, the oral cavity,feces, and aquatic habitats, as well as the hospital metagenome a term intended to encompass the genetic potential of organisms in hospitals that contribute to public health concerns such as antibiotic resistance and nosocomial infections.
Uncultured microorganisms comprise the majority of the planet earth’s biological
diversity. In many environments, as many as 99% of the microorganisms cannot be
cultured by standard techniques, and the uncultured fraction includes diverse
organisms that are only distantly related to the cultured ones. Therefore, culture independent methods are essential to understand the genetic diversity, population
structure, and ecological roles of the majority of microorganisms in a given
environmental situation. Metagenomics, or the culture-independent genomic analysis of
an assemblage of microorganisms, has potential to answer fundamental questions in
microbial ecology. It can also be applied to determining organisms which may be
important in a new industrial process still under study. Several markers have been used
in metagenomics, including 16S mRNA, and the genes encoding DNA polymerases,
because these are highly conserved (i.e., because they remain relatively unchanged in
many groups). The marker most commonly used however is the sequence of 16S mRNA.
Its potential application in biotechnology and industrial microbiology is that it can
facilitate the identification of uncultured organisms whose role in a multi-organism
environment such as sewage or the degradation of a recalcitrant chemical soil may be
hampered because of the inability to culture the organism. Indeed a method has been
patented for isolating organisms of pharmaceutical importance from uncultured
organisms in the environment.
Bioinformatics is a new and evolving science and may be defined as the use of computers
to store, compare, retrieve, analyze, predict, or simulate the composition or the structure
of the genetic macromolecules, DNA and RNA and their major product, proteins.
Important research efforts in bioinformatics include sequence alignment, gene finding,
genome assembly, protein structure alignment, protein structure prediction, prediction of
gene expression and protein-protein interactions, and the modeling of evolution.
Bioinformatics uses mathematical tools to extract useful information from a variety of
data produced by high-throughput biological techniques. Examples of successful
extraction of orderly information from a ‘forest’of seemingly chaotic information include
the assembly of high-quality DNA sequences from fragmentary ‘shotgun’DNA
sequencing, and the prediction of gene regulation with data from mRNA microarrays or
mass spectrometry.
The increased role in recent times of bioinformatics in biotechnology is due to a vast
increase in computation speed and memory storage capability, making it possible to
undertake problems unthinkable without the aid of computers. Such problems include
large-scale sequencing of genomes and management of large integrated databases over
the Internet. This improved computational capability integrated with large-scale
miniaturization of biochemical techniques such as PCR, BAC, gel electrophoresis, and
microarray chips has delivered enormous amount of genomic and proteomic data to the
researchers. The result is an explosion of data on the genome and proteome analysis
leading to many new discoveries and tools that are not possible in wet-laboratory
experiments. Thus, hundreds of microbial genomes and many eukaryotic genomes
including a cleaner draft of human genome have been sequenced raising the expectation
of better control of microorganisms. Bioinformatics has been used in the following four
areas:
a. genomics –sequencing and comparative study of genomes to identify gene and
genome functionality;
b. proteomics –identification and characterization of protein related properties and
reconstruction of metabolic and regulatory pathways;
c. cell visualization and simulation to study and model cell behavior; and
d. application to the development of drugs and anti-microbial agents.
The potential gains especially following from sequencing of the human genome and
many microorganisms are greater understanding of the genetics of microorganisms and
their subsequent improved control leading to better diagnosis of the diseases through the
use of protein biomarkers, protection against diseases using cost effective vaccines and rational drug design, and improvement in agricultural quality and quantity.
Some Contributions of Bioinformatics to Biotechnology:
Some contributions made by bioinformatics to biotechnology include automatic genome
sequencing, automatic identification of genes, identification of gene function, predicting
the 3D structure modeling and pair-wise comparison of genomes.
i. Automatic genome sequencing
The major contribution of the bioinformatics in genome sequencing has been in the: (i)
development of automated sequencing techniques that integrate the PCR or BAC based
amplification, 2D gel electrophoresis and automated reading of nucleotides, (ii) joining
the sequences of smaller fragments (contigs) together to form a complete genome
sequence, and (iii) the prediction of promoters and protein coding regions of the genome.
PCR (Polymerase Chain Reaction) or BAC (Bacterial Artificial Chromosome)-based
amplification techniques derive limited size fragments of a genome. The available
fragment sequences suffer from nucleotide reading errors, repeats –very small and very
similar fragments that fit in two or more parts of a genome, and chimera –two different
parts of the genome or artifacts caused by contamination that join end-to-end giving a
artifactual fragment. Generating multiple copies of the fragments, aligning the fragments,
and using the majority voting at the same nucleotide positions solve the nucleotide
reading error problem. Multiple experimental copies are needed to establish repeats and
chimeras. Chimeras and repeats are removed before the final assembly of the genome fragments. Using mathematical models, the fragments are joined. To join contigs, the
fragments with larger nucleotide sequence overlap are joined first.
ii. Automated Identification of Genes
After the contigs are joined, the next issue is to identify the protein coding regions or ORFs
(open reading frames) in the genomes. The identification of ORFs is based on the
principles described earlier. The two programs which are used are GLIMMER and
GenBank.
iii. Identifying gene function: searching and alignment
After identifying the ORFs, the next step is to annotate the genes with proper structure
and function. The function of the gene has been identified using popular sequence search
and pair-wise gene alignment techniques. The four most popular algorithms used for
functional annotation of the genes are BLAST, BLOSUM, ClustalX, and SMART
iv. Three-dimensional (3D) structure modeling
A protein may exist under one or more conformational states depending upon its
interaction with other proteins. Under a stable conformational state certain regions of the
protein are exposed for protein-protein or protein-DNA interactions. Since the function is
also dependent upon exposed active sites, protein function can be predicted by matching
the 3D structure of an unknown protein with the 3D structure of a known protein. With bioinformatics it is possible to predict the possible conformations of the protein coded for
by a gene and therefore the function of the protein.
v. Pair-wise genome comparison
After the identification of gene-functions, a natural step is to perform pair-wise genome
comparisons. Pair-wise genome comparison of a genome against itself provides the
details of paralogous genes –duplicated genes that have similar sequence with some
variation in function. Pairwise genome comparisons of a genome against other genomes
have been used to identify a wealth of information such as orthologous genes –
functionally equivalent genes diverged in two genomes due to speciation, different types
of gene-groups –adjacent genes that are constrained to occur in close proximity due to
their involvement in some common higher level function, lateral gene-transfer –gene
transfer from a microorganism that is evolutionary distant, gene-fusion/gene-fission,
gene-group duplication, gene-duplication, and difference analysis to identify genes
specific to a group of genomes such as pathogens, and conserved genes.
In conclusion, despite the recent emergence of bioinformatics it is already making big
impacts on biotechnology. Except for the availability of bioinformatics techniques, the
vast amount of data generated by genome sequencing projects would be unmanageable
and would not be interpreted due to the lack of expert manpower and due to the
prohibitive cost of sustaining such an effort. In the last decade bioinformatics has silently
filled in the role of cost effective data analysis. This has quickened the pace of discoveries,
the drug and vaccine design, and the design of anti-microbial agents. The major impact of
bioinformatics in microbiology and biotechnology has been in automating microbial
genome sequencing, the development of integrated databases over the Internet, and
analysis of genomes to understand gene and genome function. Programs exist for
comparing gene-pair alignments, which become the first steps to derive the gene-function
and the functionality of genomes. Using bioinformatics techniques it is now possible to
compare genomes so as to (i) identify conserved function within a genome family; (ii)
identify specific genes in a group of genomes; and (iii) model 3D structures of proteins
and docking of biochemical compounds and receptors. These have direct impact in the
development of antimicrobial agents, vaccines, and rational drug design.
SUGGESTED READINGS & REFERENCES:
1)Madigan, M., Martinko, J.M. 2006. Brock Biology of Microorganisms 11th ed. Pearson Prentice Hall,
Upper Saddle River, USA.
2) Dorrell, N., Champion, O.L., Wren, B.W. 2002. Application of DNA Microarray for Comparative
and Evolutionary Genomics In: Methods in Microbiology. Vol 33, Academic Press
Amsterdam; the Netherlands pp. 83–99.
3) Riesenfeld, C.S., Schloss, P.D., Handelsman, 2004. Metagenomics: Genomic Analysis of Microbial
Communities. Annual Review of Genetics 38, 525-52.
CITED BY KAMAL SINGH KHADKA
Assistant Professor in Pokhara University, Pokhara Bigyan Thata Prabidhi Campus, PNC, LA, NA.
Pokhara, Nepal.
Pokhara, Nepal.
Some Useful Urls:
en.wikipedia.org/wiki/Metagenomics
www.nature.com/nrmicro/focus/metagenomics/
www.cbcb.umd.edu/research/metagenomics.shtml
www.nature.com/ncomms/archive/subject/npg_subject_48/index.html
www.nature.com › Journal home › Archive › Technologies
www.nature.com/news/archive/keyword/bioinformatics.html
www.ebi.ac.uk/luscombe/docs/imia_review.pdf

Comments